NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition. Boston: Butterworths; 1990. Bookshelf ID: NBK367 PMID: 21250208
Definition
Cyanosis is a bluish color of mucous membranes and/or skin. While this is most frequently attributable to increased amounts of unoxygenated hemoglobin (deoxyhemoglobin) in the vasculature, there are other causes of bluish skin color.Technique
Daylight or artificial light sources simulating daylight's spectral composition are not necessarily optimal for detecting cyanosis. Nevertheless, consistency in observation makes it desirable that artificial light sources be similar to sunlight, since most examining areas are lighted for at least a portion of the day by sunlight. Tungsten filament bulbs and certain fluorescent bulbs are satisfactory for this purpose. If one is uncertain as to the adequacy of an artificial light source, use of sunlight will obviate this potential problem. An intense light can make cyanosis less readily apparent. One group has recommended using less than 20 footcandles of illumination. As a point of reference, the standard suggested for patient rooms in Veterans Administration hospitals is 30 footcandles, so the recommended level of light intensity to detect cyanosis is likely to be exceeded in at least some patient examination areas. When looking for cyanosis, one should inspect those body sites that contain minimal melanotic pigment, that have a capillary bed close to the skin surface, and that are well perfused. Lips, ears, trunk, nailbed, hands, conjunctiva, and circumoral areas have been compared in detecting cyanosis due to arterial hypoxemia; the tongue is the most sensitive area, but the lips are more specific.Basic Science
Blue color can be perceived in a number of situations: (1) when the light source directly shined on the retina has a predominant frequency in the upper (shorter) end of the visual spectrum; (2) when a light source with multiple frequencies (including high ones) is shined on an object, absorbing all other frequencies except those at the blue end of the visual spectrum, which are reflected to the retina; and (3) when a white light is scattered by particles, the frequencies reflected are in the high end of the visual spectrum (Tyndall effect)—the blue sky is an example of this.The normal color of flesh is thought to result from the combination of the pigments oxyhemoglobin, deoxyhemoglobin, melanin, and carotene, and from the optical effect of scattering. The importance of the latter effect has been disputed by at least one investigator, who attributes to collagen a major role in reflecting blue wavelengths. Blue skin coloration would result if the quantity of blue wavelengths reflected disproportionately increased or if the quantity of other wavelengths reflected disproportionately decreased.
Anyone who has observed a specimen of venous blood in a tube can confirm that it is not blue. Thus the blue skin color detected in individuals who have increased amounts of deoxyhemoglobin cannot be explained on the basis of reflection of increased quantities of high-frequency wavelengths from a "blue" pigment. One plausible theory to account for the observation of cyanosis under these circumstances is that deoxyhemoglobin is less red than oxyhemoglobin and therefore absorbs more red spectrum. By subtraction of red wavelengths, the blue spectrum is allowed to predominate in the reflected light (i.e., something that is less red is more blue). The bluish skin color observed with the other pigments listed in Table 45.1 is explained in a similar fashion.
According to Lundsgaard and Van Slyke (1923), as well as subsequent investigators, cyanosis generally becomes apparent when the subpapillary capillaries contain from 4 to 6 gm/dl of deoxyhemoglobin. Since this measurement was difficult to obtain directly, they proposed estimating it by averaging the amount of deoxyhemoglobin in arterial blood with that in venous blood. If one assumes a normal cardiac output, hemoglobin, and tissue extraction of O2, an arterial O2 saturation of approximately 80% would be required to cause cyanosis. It should be noted that the conclusion of Lundsgaard and Van Slyke was based on measurements of deoxyhemoglobin in peripheral venous blood and did not involve sampling of arterial blood. Their proposal of 5 gm/dl deoxyhemoglobin in mean capillary blood as a threshold for detecting cyanosis has not been confirmed or refuted by more sophisticated techniques.
Reduced arterial oxygenation can result if the amount of oxygen in the alveoli is lowered or if the gradient between the alveolar oxygen and the arterial oxygen is elevated. One can determine which of these is the explanation by measuring the arterial partial pressure of oxygen (Pao2) and calculating the alveolar partial pressure of oxygen (PAo2) and the a-a O2 gradient with the following formulas:
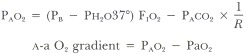
PB = barometric pressure
Ph2o37° = partial pressure water vapor at 37°C (47 mm Hg)
F1o2 = fraction of inspired air that is oxygen
PAco2 = partial pressure of carbon dioxide in arterial blood
R = respiratory quotient (Vco2/Vo2, generally about 0.8)
Even with normal arterial oxygenation, cyanosis can occur when there is increased extraction of oxygen at the capillary level because the average of arterial and venous oxygen saturation will be lower. Reduced flow through capillaries results in increased tissue extraction of oxygen (and therefore increased amounts of deoxyhemoglobin), favoring the appearance of cyanosis.
In anemic patients, much more profound decreases in tissue oxygen levels are required to produce 5 gm/dl of deoxyhemoglobin in capillary blood. For example, with a hemoglobin of 7.5 gm/dl, capillary blood would have to have a Po2 of about 19 mm Hg (33% sat.), contrasted with a Po2 of about 35 mm Hg (66% sat.) for a hemoglobin of 15 gm/dl.
Hemoglobins that have an abnormally low affinity for oxygen (high P50) have decreased amounts of hemoglobin bound with oxygen at usual levels of Pao2. Cyanosis can result on occasion.
A tube of blood containing excess methemoglobin is reddish brown to brown in color and remains so even after shaking in air or 100% O2. Methemoglobin is an oxidized hemoglobin in which iron is in the ferric form. It does not bind oxygen. Some methemoglobin is normally formed in the body, but this is usually reduced to deoxyhemoglobin by the NADH methemoglobin reductase system. If this enzyme system is deficient or if it becomes overloaded by excess amounts of methemoglobin, elevated blood levels of methemoglobin result. In some patients with congenitally abnormal hemoglobins (Hgb Ms) the structure of the hemoglobin makes the heme unit susceptible to rapid oxidation. The level of methemoglobin capable of producing cyanosis is said to be about 1.5 gm/dl, although this value seems to have been less carefully scrutinized than that for deoxyhemoglobin.
As with methemoglobin, a tube of blood containing sufficient sulfhemoglobin has a reddish brown color that does not change upon shaking in 100% O2. Sulfhemoglobin is a pigment not normally formed in the body. Its chemical composition is not well defined, although it has the spectrophotometric characteristic of strongly absorbing light at 620 nm in the presence of cyanide. The mechanism of formation is not known, although many of the same toxins that result in the oxidation of deoxyhemoglobin to methemoglobin can also produce sulfhemoglobin. The explanation for the formation of sulfhemoglobin in one individual and methemoglobin in another exposed to the same toxin is not known. Once formed, the sulfhemoglobin molecule is stable and is not converted back to deoxyhemoglobin. Cyanosis is reported to be detectable at sulfhemoglobin levels as low as 0.5 gm/dl.
Methemalbumin, which produces a brown plasma, is a pigment formed by the union of albumin in the plasma with hemin. The pigment may be present in the blood when excessive breakdown of red cells results in saturation of haptoglobin with hemoglobin. Dissolution of the remaining free hemoglobin into globin and heme can occur. Heme is immediately oxidized to hematin and in the presence of chloride forms hemin, which complexes with albumin. The minimal amount of resulting methemalbumin required to produce cyanosis is not stated in the literature.
Clinical Significance
Cyanosis as a tool for detecting arterial hypoxemia is neither sensitive nor specific. Comroe and Botelho (1947) studied a group of normal subjects breathing various concentrations of oxygen. Definite cyanosis was not apparent to 25% of observers even at arterial oxygen saturations of 71 to 75% (Pao2 35 to 40 mm Hg). In contrast, 6% and 17% of the observers believed definite cyanosis to be present when arterial oxygen saturations were 96 to 100% and 91 to 95%, respectively. In the same study progressive hypoxemia was induced in a subject and one physician first noted definite cyanosis at arterial oxygen saturations of 84%, 77%, 94%, and 82% in consecutive trials on this same subject within a period of 40 minutes. The sensitivity of this sign is lessened when examining deeply pigmented individuals. In blacks, 3 to 6% more arterial oxygen desaturation may be required for detection of cyanosis.To confirm that arterial hypoxemia is responsible for cyanosis, a blood specimen must be analyzed for Pao2. When a reduction is found, one must consider the causes listed in Table 45.2. When cyanosis is due to arterial hypoxemia, other signs and symptoms are usually present. Peripheral chemoreceptors may be stimulated by a low Pao2, causing increased ventilation with dyspnea and tachypnea. Sympathetic nervous system stimulation produces restlessness, sweating, elevation of blood pressure, and tachycardia. When hypoxemia is severe and cerebral oxygenation is impaired, confusion or coma can occur. As demonstrated in a number of studies, severe hypoxemia may be present at times when cyanosis is not readily detectable either because of observer insensitivity or confounding factors in the patient, such as heavy melanin pigmentation or anemia. The importance of arterial blood gas analysis in detecting hypoxemia cannot be overemphasized.
The usual pattern of cyanosis noted in conditions of reduced blood flow is for peripheral sites, in particular the extremities, to be affected preferentially (acrocyanosis). Central portions of the body are typically spared. Low flow may result from decreased arterial perfusion caused by poor cardiac output (as in cardiogenic shock), by fixed arterial narrowing (as in atherosclerosis), or by reflex arteriolar narrowing (as in cold weather). Venous obstruction slows capillary blood flow and may be caused by local (venous thrombosis) or central (congestive heart failure) mechanisms.
More than 20 hemoglobins with low oxygen affinities have been described. With most of them there are few clinical manifestations apart from cyanosis. Since loading hemoglobin with oxygen is not affected, whereas unloading oxygen from hemoglobin is facilitated, tissue oxygen is high, and suppression of erythropoietin may produce a mild anemia.
Deficiency of the NADH methemoglobin reductase system is inherited in an autosomal recessive pattern. Even though 15 to 25% rnethemoglobin may be present, affected individuals generally have only cyanosis as a clinical manifestation.
Oxidation of deoxyhemoglobin to form methemoglobin can be caused by many drugs and toxins including nitrites, sulfonamides, and aniline derivatives. It is unusual for symptoms to accompany the cyanosis, although, with levels of methemoglobin greater than 60 to 70%, cardiovascular collapse, coma, and death have occurred.
The five variants of Hgb M are inherited in an autosomal dominant pattern. Individuals in whom an alpha chain substitution has occurred are noted to be cyanotic beginning at birth. Those in whom beta chain substitution has occurred often do not become cyanotic until three to six months of age because of the normal changeover from gamma to beta chain synthesis during that time. Cyanosis is usually the only clinical manifestation in any of the variants.
Individuals who experience no symptoms with up to 10 gm/dl sulfhemoglobinemia have been described. Since sulfhemoglobin is stable, the rate of resolution of cyanosis following a toxic exposure is slow, as degradation of sulfhemoglobin becomes dependent on the life span of the red cell. This contrasts to cases of toxic methemoglobin formation in which the transiently overloaded NADH methemoglobin reductase system rapidly reduces methemoglobin following the removal of the toxin.
Because the formation of methemalbumin is dependent on large-scale destruction of red cells, hemolytic states or large extravascular accumulations of blood are the most common associated conditions.
Ingestion of substances containing gold or silver can produce bluish skin coloration that is most prominent in sun-exposed portions of the body. The bluish skin color associated with hemosiderin deposition is more apparent in parts of the body with less melanotic pigment. A more bronze color is seen in the presence of melanin.
Polymers of the oxidation products of chlorpromazine, when deposited in the skin and other organs, can result in a blue to purple color. A new antiarrhythmic agent, amiodarone, can cause lipofuscin deposition in the skin. In sun-exposed areas, a blue skin color is seen in a small percentage of patients on long-term therapy.
One approach to assessing the etiology of cyanosis is to obtain a heparinized arterial blood specimen. If the sample is dark red and becomes bright red on shaking in air, one should perform blood gas analysis on another specimen to confirm arterial hypoxemia. If the specimen obtained is brown and does not change color on shaking in air, the plasma should be allowed to separate. If the plasma is brown, methemalbumin is likely to be present. If the plasma is clear, one should suspect the presence of methemoglobin or sulfhemoglobin. A bright red arterial specimen obtained in a patient with generalized blue skin color should lead one to suspect deposition of nonheme pigment in the skin.
References
- *Blount SG Jr. Cyanosis: pathophysiology and differential diagnosis. Prog Cardiovasc Dis. 1971;13:595–605. [PubMed: 4933007]
- Comroe JH Jr,, Botelho AB. The unreliablity of cyanosis in the recognition of arterial anoxemia. Am J Med Sci. 1947;214:1–6. [PubMed: 20250015]
- Edwards EA, Duntley SQ. The pigments and color of living human skin. Am J Anat. 1939;65:1–33.
- Finch CA. Methemoglobinemia and sulfhemoglobinemia. N Engl J Med. 1948;239:470–78. [PubMed: 1190530]
- Geraci JE, Wood EH. The relationship of the arterial oxygen saturation to cyanosis. Med Clin North Am. 1951;35:1185–1202. [PubMed: 13098533]
- Jeghers H. Pigmentation of the skin. N Engl J Med. 1944;231:88–100. , 122–36,181–89.
- Lundsgaard C, Van Slyke DD. Cyanosis. Medicine. 1923;2:1–76.
- Mansouri A. Review: methemoglobinemia. Am J Med Sci. 1985;289:200–91. [PubMed: 4003427]
- Medd WE, French EB, Wyllie VMcA. Cyanosis as a guide to arterial oxygen desaturation. Thorax. 1959;14:247–50.
- Stadie WC. The oxygen of the arterial and venous blood in pneumonia and its relationship to cyanosis. J Exp Med. 1919;30:215–43. [PubMed: 19868355]
Tables
Table 45.1Selected Causes of Blue Skin Coloration
Increased heme pigments
|
Other pigments
|
 /
/ mismatch
mismatchTable 45.2Arterial Hypoxemia
| Mechanism | Selected etiologies |
|---|---|
| ↓ PAo2 | |
 ↓ PB ↓ PB | High altitude |
| ↓ F1o2 | Fire in a closed space |
| ↑ Paco2 (hypoventilation) | Depression of respiratory center (narcotics, trauma) |
| Anterior horn cell disease (ALS polio) | |
| Neuromuscular junction disease (myesthenia gravis, botulism) | |
| Muscle disorder (muscular dystrophy, muscle fatigue) | |
| Chest wall disease (kyphoscoliosis) | |
| ↑ a-a O2 gradient | |
| Right-to-left shunt | |
| Intracardiac | Cyanotic congenital heart disease (tetralogy of Fallol) |
| Intrapulmonary | |
| Congenital | Osler-Weber-Rendu disease |
| Acquired | Cirrhosis |
| Physiological | ARDS |
| &Vdot;/&Qdot; mismatch | COPD |
| Diffusion defect | Rare cause of significant hypoxemia |
Copyright © 1990, Butterworth Publishers, a division of Reed Publishing.
Không có nhận xét nào:
Đăng nhận xét